
��(d��ng)ǰλ�� > ��� > ���g(sh��)���� > ���ڙz�y�Ͷ��������O�Լ�(x��)�����x���һ�N�·f��HILIC LC-MS����
���ڙz�y�Ͷ��������O�Լ�(x��)�����x���һ�N�·f��HILIC LC-MS����
���ڙz�y�Ͷ��������O�Լ�(x��)�����x���һ�N�·f��HILIC LC-MS����
Henry Shion1��John Shockcor1��Evan Bernier2��Stephen Mcdonald2��Kirsten Hartil3��Bhavapriya Vaitheesvaran3��Haitao Lu3��Gustavo Palacios3��Inwin J. Kurland3��Alan L. Millar1
1��������˾�������R�_�T�����נ�������2��������˾�������R�_�T����ؐ������3�q̫��W(xu��)��������Ÿ����˹̹�t(y��)�W(xu��)Ժ�������~�s�в��ʿ�˹�^(q��)
����
��(f��)�s�����Ʒͨ�����������ɷ�ӳ�ЙC(j��)�w���x��B(t��i)�ă�(n��i)Դ�Դ��x�����e�ǹ������ɸ���(j��)���ǿ��տs������ɫ�������ǽͽ��ͣ�߀�����տs�����tɫ�����������ͣ������F(xi��n)�������ԵĴ��x�M��ָ�y�D�V�����������tɫ���⑪(y��ng)�@ʾһ�N���������w���x���������ѭ�h(hu��n)���Ĵ��x�Mָ�y�D�V������ɫ���⑪(y��ng)�@ʾ�����ǽͽ���x��Ĵ��x�Mָ�y�D�V���侀���w���x��ĝ�ȴ����ڼtɫ���⡣�mȻ�ܶ��@��x��ɾ����^��(qi��ng)�ĘO�������@Щ�ӱ�ͨ��ʹ�÷���Һ��ɫ�V-�|(zh��)�V��RP-LC-MS���M(j��n)�з������ɴ˶����Ķ����^��ı������ڱ��о������҂��_�l(f��)��һ�N���ڙz�y�Ͷ�����(qi��ng)�O�Լ�(x��)�����x����·fLC-MS���������о������˻��ځ�2���w��BEH���������Hˮ����Һ��ɫ�V��HILIC������(li��n)�����s���ĘO�U�w�Еr(sh��)�g�|(zh��)�V�x�������ĘO�U�|(zh��)�V��
����
�@Щԇ�(y��n)����(li��n)����������SynaptTM HDMSTM��������Synapt G2 HDMS��Xevo TQ��һ�_UPLCϵ�y(t��ng)���M(j��n)�У�
Һ��ɫ�V�l����1����ʹ�ÉA��������
Һ��ɫ�Vϵ�y(t��ng)��������ACQUITY UPLC®ϵ�y(t��ng)
�� ɫ�V����ACQUITY UPLC BEH Amide ����������2.1��100mm��1.7µm
�� ���أ�45��
�� ���٣�500µL�����
�� �A��������A�����棯ˮ��95/5��v/v����10mM�����@��pH=9.0
�� �A��������B�����棯ˮ��50/50��v/v����10mM�����@��pH=9.0
�� �ݶȣ����ÉA���������M(j��n)�з��x�r(sh��)���������ݶ�������2% B - 85% BϴÓ8��犣�Ȼ����98% B��ˮƽ�±���2������������ص�2% B��ˮƽ�M(j��n)����ƽ�⣨5��犣�
�� �M(j��n)������5-20µL
Һ��ɫ�V�l����2����ʹ������������
Һ��ɫ�Vϵ�y(t��ng)��������®ACQUITY UPLC®ϵ�y(t��ng)
�� ɫ�V����ACQUITY UPLC BEH Amide ����������2.1��100mm��1.7µm
�� ���أ�40��
�� ���٣�600µL�����
�� ����������A�����棯ˮ��95/5��v/v����10mM�����@��pH=3.0
�� ����������B��ˮ��10mM�����@��pH=3.0
�� �ݶȣ�����5% B - 30% BϴÓ5������ڵ�5.5min�r(sh��)����60% B������7��犣��������ص�5% B��ˮƽ�M(j��n)����ƽ�⣨5��犣�
�� �M(j��n)������20µL�M�����h(hu��n)
�|(zh��)�V����
Synapt HDMS��Synapt G2 HDMS
ԓ�|(zh��)�V�x�����x��MSEģʽ���\(y��n)�������õ�ë��(x��)��늉���3.0kV��Դ�ضȺ�ȥ�܄����ضȷքe���O(sh��)����120���400����
��MSE�ɼ�ģʽ�£��x�������ڵ�������ײ������B(t��i)���M(j��n)�н���������@�ӿ���һ�η�����(n��i)ͬ�r(sh��)�ռ����з������ĸ�x�Ӻ���Ƭ�x�ӵ���Ϣ���������T��DDA��������Ҏ(gu��)�����������IJɘ�ƫ�����ڳ�Ҏ(gu��)���������ض���m/z�������Ƭ��ǰ�����x������
Xevo-TQ�|(zh��)�V
Xevo TQ��ͬ�r(sh��)�����x�Ӻ�ؓ(f��)�x��ģʽ���\(y��n)����ë��(x��)��늉���3.0kV��Դ�ضȺ�ȥ�܄����ضȷքe���O(sh��)����150���650����ȥ�܄������w���ٱ��O(sh��)����1200L��С�r(sh��)����ײ���w���壩�����ٞ�0.18mL����犣�4��10-3���ͣ���MS1/MS2�ֱ��ʱ��O(sh��)�Þ��λ�|(zh��)����0.75Da FWHM����
�M����ȡ��Ę�Ʒ�Ƃ�
�tɫ�ı�Ŀ�~����S���Á��c��ɫ�����c����G���M(j��n)�б��^������Ľ�ʳһ��ҹ��BALB/cС�������²��M(j��n)���˼�����������x��ͨ�^�M�����|(zh��)���ķ�����1:1�ļ״���ˮ����܄����M(j��n)����ȡ��֬�|(zh��)ͨ�^���ȷ���1:1�ı����������״���ˮ��ȡҺ�ж��õ�ȥ����ȥ֬�|(zh��)��ĽM����ȡ�ӱ��քe��400µL��4000µL��90:10���棺ˮ����܄��M(j��n)�Г]�ɏ�(f��)�ܡ�����ijЩ�M�ֵĺ������ڶ������������Ҳ��ÿ�ݘӱ���ϡ�10���M(j��n)���˷�����
�Y(ji��)����ӑՓ
1. ͨ�^(li��n)��Synapt HDMS��UPLC�M(j��n)��HILIC LC-MS�����_�l(f��)
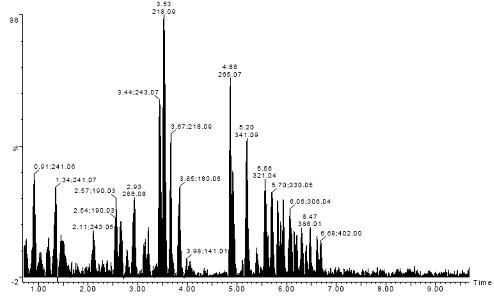
�D1. 108�N��(bi��o)��(zh��n)��(qi��ng)�O�Լ�(x��)�����x��Ŀ��x��ɫ�V�D�����а��������ᡢDNA/RNA�A�����������}���oøA��B��S���ء��ЙC(j��)�����
�D1�o����108�N��(bi��o)��(zh��n)��(qi��ng)�O�Լ�(x��)�����x��Ŀ��x��ɫ�V�D��TIC�������а��������ᡢDNA/RNA�A�����������}���oøA��B��S�������ЙC(j��)��ȡ����(xi��ng)ԇ�(y��n)ͨ�^(li��n)��������Synapt HDMS�����x��MSEģʽ�£���������UPLC��Һ��ɫ�V�l����1�������M(j��n)����
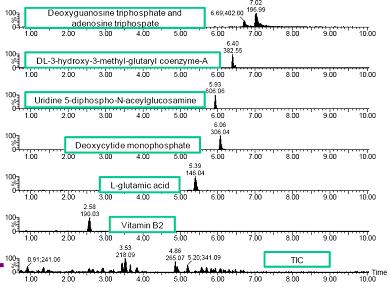
�D2. ��һ��������ȡ�x��ɫ�V�Dʾ����50mDa���ڃ�(n��i)��
�D2�o�����P(gu��n)�چ�һ��������ȡ�x��ɫ�V�DƬ�Σ�50mDa���ڣ���һЩ���ӡ����xȡ����ȡ�x��ɫ�V�D�еĴַ匒�y��ֵ����5-15��֮�g��Ȼ����Ҳ�^�쵽����β������匒�L�_(d��)60����������҂����]��Һ��ɫ�V�ėl����
2. ʹ��UPLC��Xevo-TQ(li��n)�Ãx����HILIC LC-MS�����_�l(f��)��(bi��o)��(zh��n)Ʒ�ͼ�(x��)����ȡ���M(j��n)�ж�������
��UPLC��(y��u)���^�����M(j��n)�еĘ�(bi��o)��(zh��n)Ʒ��������ʹ������pH�������ྏ�_Һ����ߌ���������ЙC(j��)��ķֱ���������Ⱥͷ匦�Q�ԡ��D3���^�ˌ�һ�N�����s2µg/mL L-ه����ļ���������Һ���ó��ăɷ�MRMɫ�V�D�����ϵ�ɫ�V�Dͨ�^ʹ�ÉA�������ࣨҺ��ɫ�V�l��1�����M(j��n)�еķ��x���ó��������µ�ɫ�V�D�t�������������ࣨҺ��ɫ�V�l��2����
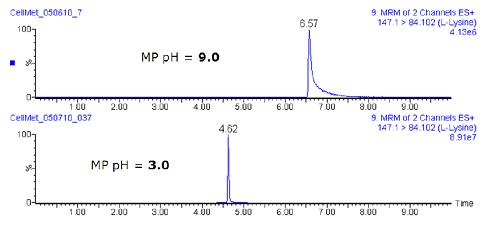
�D3. ���^��ͬpH�£������s2µg/mL L-ه������Һ�ăɷ�MRMɫ�V�D
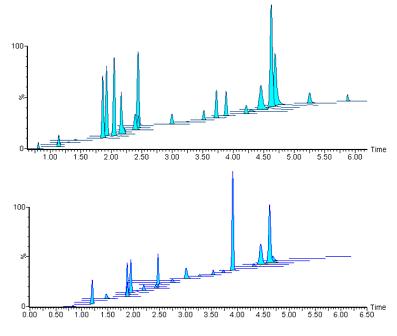
�D4. Һ��ɫ�V�l����2���ó���MRMɫ�V�Dʾ��
�D4�����������ࣨҺ��ɫ�V�l����2������1600ng/mLУ��(zh��n)��(bi��o)��(zh��n)Ʒ���ψD���͘ӱ�H-S��0090����MRMɫ�V�D�M(j��n)���˯B�ӡ�
�ڴ˗l�������҂��ɹ����x������������28�N��������а������������ЙC(j��)��������}���D6����
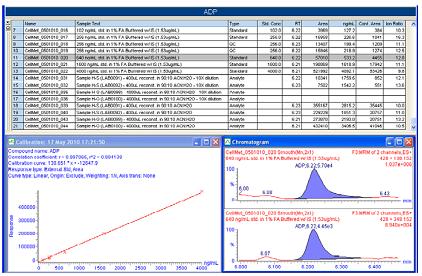
�D5. TargetLynx�o����ADP��ӈ�(b��o)��ʾ��
43�N������Ķ����������Ȱ�������Ҳ�����A�ԗl����ʹ��TargetLynxTM�M(j��n)��������ÿ�N�����������������(y��ng)����ӈ�(b��o)����Ӌ(j��)��Y(ji��)��ͨ�^���Իؚw�M(j��n)�д_�����D5�����ն����ᣨADP����������ѭ�h(hu��n)���e��Ҫ����һ��(g��)��(b��o)��ʾ����
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
�D6. ʹ�ÉA�ԣ�����������ԣ��ұ�����������H-S��H-G�M���ӱ������z��������ĝ��Ӌ(j��)��Y(ji��)������
���ډA�Ժ�������������x�����z����H-S��H-G�M���ӱ��е�ÿ�N�������Ӌ(j��)���˷���������ͨ�^���^�tɫ��Ŀ�~����S�������c����G���M���ӱ��еĴ��x�����C��(sh��)������������ȷ�������@�����
3. ͨ�^(li��n)��Synapt G2��UPLC��HILIC LC-MS�����Ę�(bi��o)��(zh��n)Ʒ�ͼ�(x��)����ȡ���M(j��n)�ж�������
����_�l(f��)������Synapt G2��QuanTof���g(sh��)[1]�Լ�MSE��(sh��)��(j��)�ɼ����g(sh��)�đ�(y��ng)�Ì�(sh��)�F(xi��n)��һ�Nͨ����UPLC-MS�������_�l(f��)���@�N��������һ���\(y��n)�з����õ��ṩ�������|(zh��)�Ķ����Ͷ�����Ϣ���Ȱ���ĸ�x����ϢҲ�����a(ch��n)���x�ӵ���Ϣ�����ڴ��(xi��ng)�y��������ϵ��ϡጘ�(bi��o)��(zh��n)Ʒ�Լ�H-S��H-G�ӱ��Ĕ�(sh��)��(j��)���M(j��n)���˲ɼ����D7�������еĔ�(sh��)��(j��)̎�������ڲɼ���ɺ��M(j��n)����
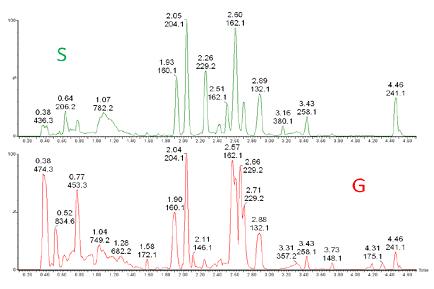
�D7. ȫ����ɫ�V�D���^��ͨ�^UPLC-Synapt G2�@ȡ��S��G��(x��)����ȡ��Ĕ�(sh��)��(j��)��
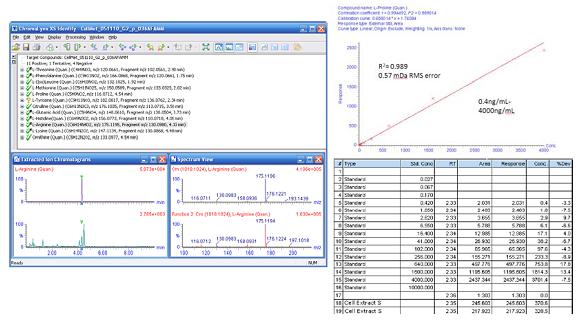
�D8. Ŀ��(bi��o)��������b���Ͷ����Y(ji��)��
�������ԄӺY�x��(x��)����ȡ�����Ƿ����Ŀ��(bi��o)���������x�ӵ��b���ʹ_�J(r��n)ͨ�^һ��(g��)��һ̎�����Eʹ��Posit��veTMܛ�����ó�ĸ�x�Ӻ����x�ӵĜ�(zh��n)�_�|(zh��)����2mDa���ڣ�����(sh��)�F(xi��n)���D8����
ԓ��(sh��)��(j��)�����cXevo TQ�����Y(ji��)���������õ�һ�������D8�o����һ��(g��)ʾ����������L(f��ng)-�������ڼ�(x��)����ȡ���б����_�b�e����������4��(g��)��(sh��)�����б��F(xi��n)�����ԵĄӑB(t��i)����푑�(y��ng)��RMS�|(zh��)���`���0.57mDa��
�Y(ji��)Փ
ʹ������BEH Amide���������_�l(f��)��һ�N���ڷ��x�O�ԽM����(x��)�����x���HILIC LC-MS���������\(y��n)�Еr(sh��)�g����15�����
ͬ�r(sh��)����Xevo TQ��Synapt G2 HDMS���tɫ�Ͱ�ɫ�ļ���M����ȡ���M(j��n)�ж��������������@�^(q��)�ֳ�����Ҫ���x�����AMP��ADP��ATP���Լ��ϰ��ᣩ���@����ͬ��
QuanTof���g(sh��)��(sh��)�F(xi��n)�˶��ԺͶ���ԇ�(y��n)?z��i)܉���Synapt G2��ͬ�r(sh��)�M(j��n)�С�
�����īI(xi��n)
1. Wildgoose J�������˻���ADC�·f�z�y���g(sh��)�ĸ�Ч�����ۯB��OA-Tof �|(zh��)�������x����18�Ç��H�|(zh��)�V�����IMSC�����ύ�Ĺ�����������÷��2009����
��Դ���������Ƽ����Ϻ�������˾
(li��n)ϵ�Ԓ��800(400)8202676 (���M(f��i)�Ԓ֧����)
E-mail��[email protected]
(li��n)ϵ�Ԓ��800(400)8202676 (���M(f��i)�Ԓ֧����)
E-mail��[email protected]

- ICP-MS�y�������ͳ��e�����ؽ��ٵIJ�ͬǰ̎�����������о�
- AFADESI-Orbitrap�������Y���x�C(j��)���о������پ���(zh��n)�t(y��)�W(xu��)
- ِĬ�w�o�C(j��)�|(zh��)�V���g(sh��)�ڜy��ͬλ�ر�ֵ�еđ�(y��ng)��
- (li��n)���|(zh��)�V����������ˎ�|(zh��)�����������˼�늺ɮ���(g��u)�w�Ƃ���ͻ��
- �x�����|(zh��)�V���������ȹ��w�o�C(j��)��Ʒ�Ŀ��ٺ���Ԫ�ط���
- ِĬ�wStellar�|(zh��)�V�x��Ѫ�{�����|(zh��)�M�W(xu��)�еđ�(y��ng)��
- ِĬ�w�Fˎ�Y��߷ֱ�ɫ�V�|(zh��)�V������������ʳƷ��ȫ
- ِĬ�wTSQ Plusϵ��Һ�|(zh��)(li��n)�Ãx�������ļ�����ˎ�ﶨ������
- SCIEXȫ���ò̿���:��(d��ng)�|(zh��)�V���^�Q����չ���A���a(ch��n)
- ���݂��c�ϷʾC���ԇ��ҿƌW(xu��)���ĭh(hu��n)���о�Ժ
- ���݂�չʾȫ��Infinity III������(zh��n)���M(j��n)չ
- �������c����ǻ۾G��������Ⱦ��z�y(li��n)�ό�(sh��)�(y��n)��
- 2024Ľ����Ϻ���������չ����ʢ���_Ļ
- 2024Ľ����Ϻ���������չ��չ��Get�öY
- 2024Ľ����Ϻ���������չ�M�F(tu��n)��(b��o)�����1�쵹Ӌ(j��)�r(sh��)
- CHINA LAB 2025�V�ݷ����yԇ����(sh��)�(y��n)���O(sh��)��չ֪ͨ


Copyright(C) 1998-2025 �������ľW(w��ng) �Ԓ��021-64166852;13621656896 E-mail��[email protected]